
"I tell you, it is tough on the family," says Felix Bidemi, a psychiatric nurse who lives in Athy, Co Kildate, "Physically, emotionally, mentally and financially, it is so tough on the family." Two of Bidemi's children have sickle cell anaemia, an inherited blood disorder that can have life-threatening complications.
When both parents carry the sickle cell gene, their child has a one-in-four chance of having sickle cell anaemia, the most common type of sickle cell disease (SCD). It particularly affects people of African descent, and World Sickle Cell Awareness Day is Friday, June 19th.
Bidemi describes the two boys, aged nine and seven, as “great warriors” in their battle with this challenging condition. In the older boy, signs emerged at about six months of age. “He wouldn’t sleep, he would be crying. I didn’t have the knowledge of sickle cell disease. I was just thinking, ‘why is this boy crying’? One night he did not sleep all night and we decided to call an ambulance.”
At the hospital, SCD was first mentioned. The infant’s hands and legs were swollen and he was put on pain relief.
The diagnosis was later confirmed.
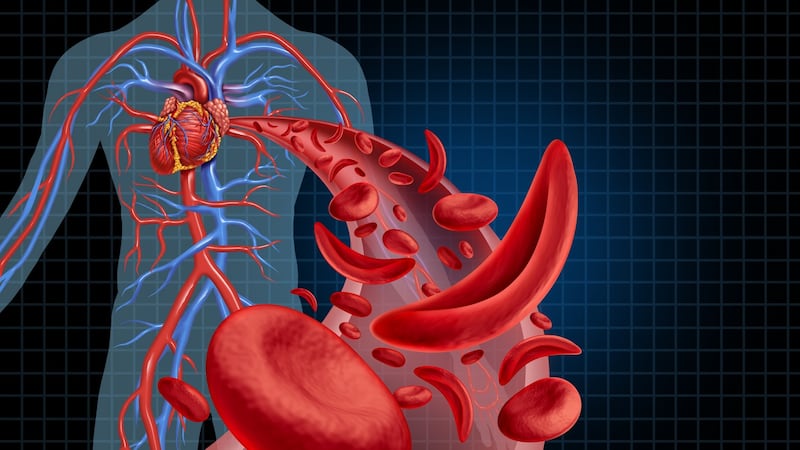
SCD is so-named because abnormal haemoglobin – the protein that transports oxygen in the blood – causes red blood cells to become rigid, sticky and shaped like sickles. These atypical cells have a shorter life span and get stuck in blood vessels, slowing or blocking blood flow and oxygen to parts of the body. Pain crises are a clinical hallmark of the condition and can be excruciating, often requiring emergency department attendance. Serious complications include stroke and other organ damage, but treatment advances have improved life expectancy. People can have a good quality of life into middle age and beyond.
As well as following medication regimens, Bidemi’s two boys take precautions such as keeping hydrated, avoiding strenuous activity, and staying home from school if a bug is circulating – something the rest of the country can now understand.
But a sickle cell crisis could occur despite preventative measures and the family remain vigilant.
Every five weeks, the boys attend Children's Health Ireland (CHI) at Crumlin for an exchange blood transfusion to lower the risk of complications. "They do find it hard, but as they are growing up, at times they do ask questions, 'Daddy why do we have to go to hospital every time?' We have started explaining to them what type of sickness they have and the treatment they must have to make them function."
As people with SCD are vulnerable to infections, Covid-19 presents an additional concern. There are posters up in the family home “just to draw their awareness to it, ‘look, you have to do your hand-washing’” explained Bidemi, “’We practise social distancing in the house and take precautions.”
Increase in cases in Ireland
In the early 2000s, as Ireland became more multi-ethnic, cases of SCD rose significantly. Paediatric haematologist Prof Corrina McMahon recalls “a prophesy” in parts of the health service that the issue would “disappear” and wouldn’t be “a big deal”.

Prof McMahon leads the haemoglobinopathy service at Crumlin, which is accessed by around 360 children and young adults with SCD. Approximately 163 adults with SCD are attending St James’s Hospital, Dublin. Both services have smaller cohorts with thalassaemia, another inherited blood disorder.
The SCD programme includes hydroxyurea (a chemotherapy drug) and blood transfusion clinics, as well as assessments using MRI and transcranial doppler ultrasound (measuring blood flow in the brain).
Two specialist nurses work with Prof McMahon, who receives tertiary referrals for red blood cell and haemoglobin disorders in neonates and children nationally.
This level of consultant manpower is far short of a UK minimum recommendation of one consultant for every 200 patients with SCD. Recently, the hospital advertised for an additional consultant in non-malignant haematology.
The SCD service is not designated as a national centre and it does not have ring-fenced funding. It shares a psychologist, social worker and physiotherapist, while a neonatal screening programme runs on an unfunded basis with maternity hospitals.
Preventative strategies had helped to ensure no case of stroke for approximately five years. More than 100 patients were on transfusion programmes and about 150 patients on hydroxyurea therapy, and a significant number were not on disease-modifying medication. Managing these strategies entailed huge organisation and commitment from the small clinical team.
Even with the best will in the world, people die, and young people die
Prof McMahon believes a national clinical programme for haemoglobinopathy is required to drive improvements, capacity and cohesion nationally. “The reality is that sickle is a life-threatening disorder and even with the best will in the world, people die, and young people die,” says the consultant, who emphasised that everything possible must be done to reduce this risk.
The transition between paediatric and adult services for SCD patients is recognised as a risk period and managed “carefully” between Crumlin and St James’s.
Patients now stay with Crumlin until after their first year at college (or equivalent). However, if a patient outside Dublin turns 17 and suddenly must present to local adult services - where SCD protocols may not be as established as in the local paediatric department - this could be detrimental to their management. “There needs to be a national way of doing things…to actually engage in a conversation to see what works,” says Prof McMahon.
Lack of access to stem cell transplant is a concern. This procedure has risks and requires a matched donor but is the only curative therapy. No SCD patients have accessed this procedure abroad through the HSE Treatment Abroad Scheme (TAS) since 2012, according to Prof McMahon.
Patients are required to stay near the transplant centre for three months post-procedure for close monitoring and the TAS does not cover accommodation costs.
CHI at Crumlin was finalising an agreement with a hospital in northern Italy to facilitate five transplants per year, with low-cost accommodation available, but the Covid-19 pandemic has stalled this process.
Crumlin has a “beautiful transplant unit” and it would make “far more sense” to undertake these transplants at the hospital, says Prof McMahon. A CHI spokesperson said there are capacity constraints in facilitating transplant for non-malignant conditions such as SCD.
When you have knowledge and hear about something that can cure a sickness or disease, you want to go for it
Bidemi says he has raised the matter of stem cell transplants at Crumlin. “When you have knowledge and hear about something that can cure a sickness or disease, you want to go for it, that is what we are asking,” he says.
He would like to see more funding for SCD research and advises that healthcare professionals should not assume people are drug-seeking, which is a noted stigma internationally. SCD is not just an “immigrant problem” with many affected patients being Irish citizens, he added.

Esther Pepple Onolememen, founder of Sickle Cell Society Ireland, has been collaborating with the Royal College of Surgeons in Ireland to deliver information sessions to medical students in an "early patient contact" module.
Onolememen, a social worker who has two children with SCD, believes this will make a “huge difference” to the knowledge of future healthcare professionals. “If every college could adopt this module that would be fantastic.”
Lack of awareness, even within the health sector, is still an issue.
A family support service in the community is needed to assist with psycho-social challenges that impact on health and wellbeing for SCD patients and families, says Onolememen.
This point is also raised by the Sickle Cell and Thalassaemia Ireland group. It said education on the genetic inheritance aspect of SCD and thalassaemia is “very important”.
Adult patients
While SCD has a variable course for individuals, management becomes more complex as patients age.
Consultant haematologist Dr Emma Tuohy, who leads the adult haemoglobinopathy service at St James's Hospital, says: "Patients have different phenotypes of their disease but many patients do eventually get end-organ damage and then if you add in the other risk factors – patients getting older, more hypertension, more diabetes, we do find these patients do get sicker as they get older."
The adult service, which has been developing since 2015, has a young cohort. About 70 per cent of patients are under 26 years of age. It has two clinical nurse specialists and shared allied health professionals.
It is expected that a further 120 patients will access the adult service within the next three years.
With demands set to grow “very rapidly”, there are major capacity constraints.
The service currently does not have the physical space capacity to provide blood transfusion to its future cohort of patients who need to transition from Crumlin. This is critical as transfusion is part of the standard of care.
It is really about trying to get the resources and the staffing
In addition to capacity, more staffing will be required to support best-practice care and a funding submission has been made to the HSE. The hospital has funded an extension to provide more space in the shorter term, and this was about to proceed before the Covid-19 crisis erupted. Work will begin in the coming weeks. “My colleagues in St James’s are fantastic, we have a great multidisciplinary service, so it is the best hospital from my perspective to have this cohort of patients. It is really about trying to get the resources and the staffing.”
There are “high achieving patients” progressing well in their education and employment, says Dr Tuohy.
Sadly, there has been one death in the adult cohort, which occurred at another hospital in Ireland, and three children with SCD have died in the last five years.
Dr Tuohy says this highlighted that SCD patients incur significant morbidity and mortality. She says it is essential that both sites are recognised as national centres, and have a national clinical lead who can implement national guidelines along with a framework as to how this cohort of patients should be managed.
Effects of stress
Difficulties associated with migration can be an added stress on people with SCD, and their loved ones, and this is true for an Dundalk-based family.
Ayodeji Adesanya's daughter Janet (19) has SCD. In 2016, Adesanya returned to Ireland with her children, having lived here for a short period in the early 2000s. She applied for residency (on the basis of having an Irish citizen child) but it was two years before it was granted. The family stayed in emergency accommodation during this period. "There was a time when four of us were just staying in one room," she says. "I think [Janet] was 17 then, she would sleep on a bed with her 15-year-old brother… That was in emergency accommodation in Drogheda. "

Factors such as stress and cold temperatures can trigger complications in SCD, therefore living circumstances have particular importance.
It may not be well understood that SCD can be life-threatening, says Adesanya. “It is not something that is common here, I understand that, but maybe with more enlightenment or education, people will realise they are really vulnerable people… One moment people can be smiling and playing - the next one, they have the crisis,” says Adesanya, who has retrained in Ireland as a healthcare support worker.
Adesanya is thankful her daughter receives good clinical care here. “I really don’t want to think what would have happened if we were still back home [in Nigeria].”
Commenting on her experience of SCD, Janet says: “I have to go into the hospital once a month to get a blood exchange – they put what is called a port-a-cath in my chest, and I take medications as well, all types of medication.”
Janet has to be careful to not do anything too physically strenuous; she says having SCD “has just been really hard”.
Janet says her favourite subject in school was English and she would like to further this interest.
However her immigration stamp – stamp 3 – presents barriers to accessing employment and third-level programmes. She has completed a PLC course in the meantime. The family is hoping for a solution.
“She wants to be a writer,” says Adesanya. “She has been writing a lot of things. She went in for pre-Arts. That is what she wanted to do in college.”

















